

Wilson Disease
Return to Overview PageDefinition & Facts
In this section:
- What is Wilson disease?
- How common is Wilson disease?
- Who is more likely to have Wilson disease?
- What are the complications of Wilson disease?
What is Wilson disease?
Wilson disease is a genetic disorder that prevents the body from removing extra copper, causing copper to build up in the liver, brain, eyes, and other organs.
Your body needs a small amount of copper from food to stay healthy, but too much copper is harmful. Without treatment, Wilson disease can lead to high copper levels that cause life-threatening organ damage.
How common is Wilson disease?
Experts are still studying how common Wilson disease is. Older studies suggested that about 1 in 30,000 people have Wilson disease.1 These studies were conducted before researchers discovered the gene mutations that cause Wilson disease.
Newer studies of people’s genes suggest that Wilson disease may be more common. A study in the United Kingdom found that about 1 in 7,000 people have gene mutations that cause Wilson disease.2
Experts aren’t sure why gene studies suggest that Wilson disease is more common than previously thought. One reason might be that some people with Wilson disease are not diagnosed. Another reason might be that some people have gene mutations for Wilson disease but don’t develop the disease.
Who is more likely to have Wilson disease?
People have a higher chance of having Wilson disease if they have a family history of Wilson disease, especially if a first-degree relative—a parent, sibling, or child—has the disease.
People who have Wilson disease typically develop symptoms when they are between ages 5 and 40.3 However, some people develop symptoms at younger or older ages. Doctors have found the first symptoms of Wilson disease in infants as young as 9 months and in adults older than 70 years.1,4
What are the complications of Wilson disease?
Wilson disease may lead to complications, but early diagnosis and treatment can lower your chances of developing them.
Acute liver failure
Wilson disease can cause acute liver failure, a condition in which your liver fails rapidly without warning. About 5 percent of people with Wilson disease have acute liver failure when they are first diagnosed.5 Acute liver failure most often requires a liver transplant.
Acute kidney failure and a type of anemia called hemolytic anemia often occur in people who have acute liver failure due to Wilson disease.
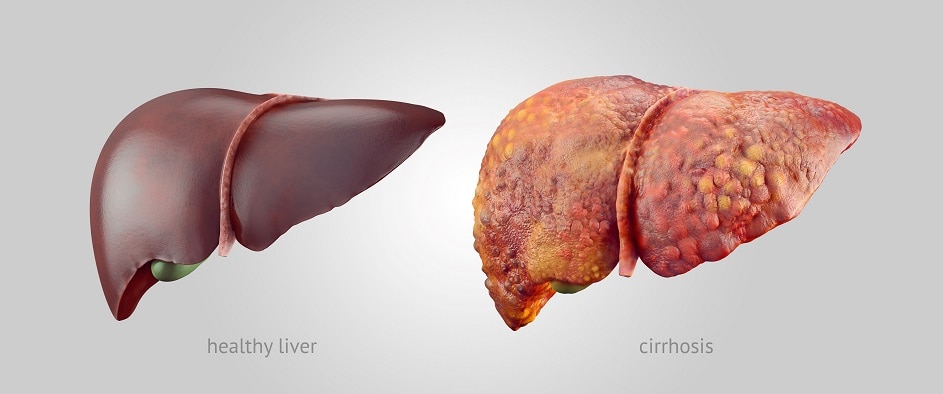
Cirrhosis
In cirrhosis, scar tissue replaces healthy liver tissue and prevents your liver from working normally. Scar tissue also partly blocks the flow of blood through the liver. As cirrhosis gets worse, the liver begins to fail.
Among people who are diagnosed with Wilson disease, 35 to 45 percent already have cirrhosis at the time of diagnosis.6
Cirrhosis increases your chance of getting liver cancer. However, doctors have found that liver cancer is less common in people who have cirrhosis due to Wilson disease than in people who have cirrhosis due to other causes.
Liver failure
Cirrhosis may eventually lead to liver failure. With liver failure, your liver is badly damaged and stops working. Liver failure is also called end-stage liver disease. This condition may require a liver transplant.
References
Symptoms & Causes
What are the symptoms of Wilson disease?
The symptoms of Wilson disease vary. Wilson disease is present at birth, but the symptoms don’t appear until the copper builds up in the liver, the brain, or other organs.
Some people do not have symptoms of Wilson disease before they are diagnosed with the disease and treated. If you do have symptoms, the symptoms may be related to your liver, nervous system and mental health, eyes, or other organs.
Liver symptoms
People with Wilson disease may develop symptoms of hepatitis, or inflammation of the liver. In some cases, people develop these symptoms when they have acute liver failure. These symptoms may include
- feeling tired
- nausea and vomiting
- poor appetite
- pain over the liver, in the upper part of the abdomen
- darkening of the color of urine
- lightening of the color of stool
- yellowish tint to the whites of the eyes and skin, called jaundice
Some people with Wilson disease have symptoms only if they develop chronic liver disease and complications from cirrhosis. These symptoms may include
- feeling tired or weak
- losing weight without trying
- bloating from a buildup of fluid in the abdomen, called ascites
- swelling of the lower legs, ankles, or feet, called edema
- itchy skin
- jaundice
Nervous system and mental health symptoms
People with Wilson disease may develop nervous system and mental health symptoms after copper builds up in their body. These symptoms are more common in adults but sometimes occur in children.7 Nervous system symptoms may include
- problems with speech, swallowing, or physical coordination
- stiff muscles
- tremors or uncontrolled movements
Mental health symptoms may include
- anxiety
- changes in mood, personality, or behavior
- depression
- psychosis
Eye symptoms
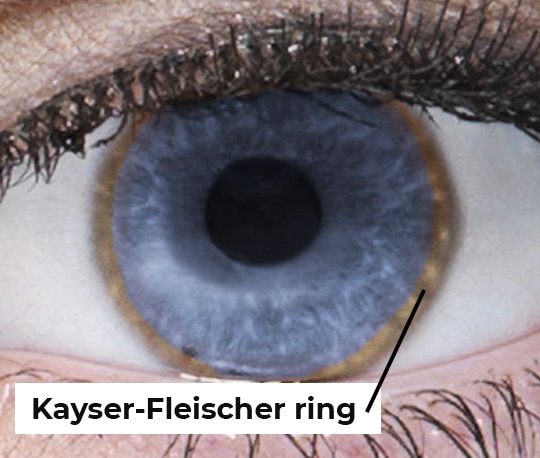
Many people with Wilson disease have Kayser-Fleischer rings, which are greenish, gold, or brownish rings around the edge of the corneas. A buildup of copper in the eyes causes Kayser-Fleischer rings. A doctor can see these rings during a special eye exam called a slit-lamp exam.
Among people who have nervous system symptoms of Wilson disease, more than 9 out of 10 have Kayser-Fleischer rings. However, among people who have only liver symptoms, 5 or 6 out of 10 have Kayser-Fleischer rings.7
Source: Obtained from the Wilson Disease Association
Other symptoms and health problems
Wilson disease can affect other parts of your body and cause symptoms or health problems, including
- a type of anemia called hemolytic anemia
- bone and joint problems, such as arthritis or osteoporosis
- heart problems, such as cardiomyopathy
- kidney problems, such as renal tubular acidosis and kidney stones
What causes Wilson disease?
Mutations of a gene called ATP7B cause Wilson disease. These gene mutations prevent the body from removing extra copper. Normally, the liver releases extra copper into bile. Bile carries the copper, along with other toxins and waste products, out of the body through the digestive tract. In Wilson disease, the liver releases less copper into bile, and extra copper stays in the body.
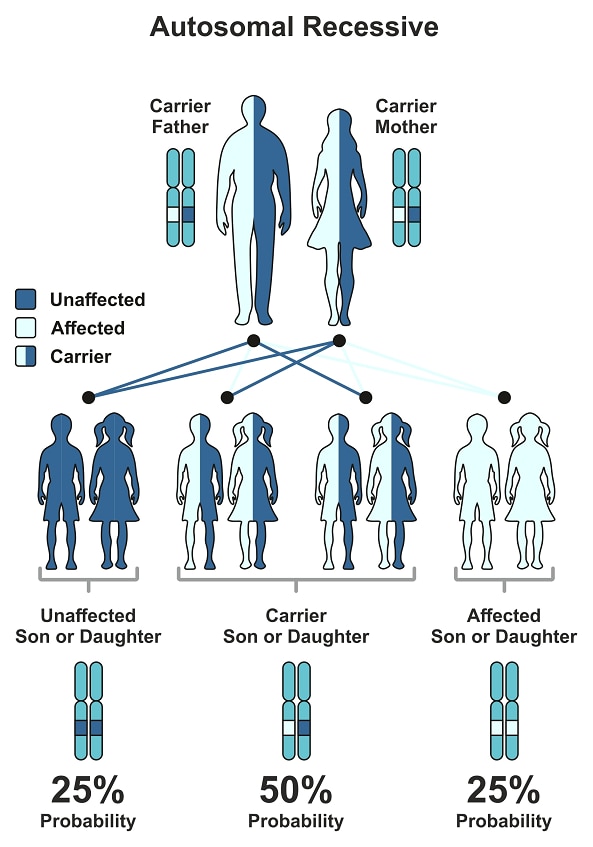
The ATP7B mutations that cause Wilson disease are inherited, meaning they are passed from parent to child. These mutations are autosomal recessive, meaning that a person must inherit two ATP7B genes with mutations, one from each parent, to have Wilson disease. People who have one ATP7B gene without a mutation and one ATP7B gene with a mutation do not have Wilson disease, but they are carriers of the disease.
People can inherent Wilson disease if both parents are carriers who don’t have the disease. The chart below shows the chance of inheriting Wilson disease from parents who are carriers.
References
Diagnosis
How do doctors diagnose Wilson disease?
Doctors diagnose Wilson disease based on your medical and family history, a physical exam, an eye exam, and tests.
Medical and family history
Your doctor will ask about your family and personal medical history of Wilson disease and other conditions that could be causing your symptoms.
Physical exam
During a physical exam, your doctor will check for signs of liver damage such as
- changes in the skin
- enlargement of the liver or spleen
- tenderness or swelling in the abdomen
- swelling in the lower legs, feet, or ankles, called edema
- yellowish color of the whites of the eyes
Eye exam
During a slit-lamp exam, a doctor will use a special light to look for Kayser-Fleischer rings in your eyes.

What tests do doctors use to diagnose Wilson disease?
Doctors typically use blood tests and a 24-hour urine collection test to diagnose Wilson disease. Doctors may also use a liver biopsy and imaging tests.
Blood tests
For a blood test, a health care professional will take a blood sample from you and send the sample to a lab.
Your doctor may order one or more blood tests, including tests that check amounts of
- ceruloplasmin, a protein that carries copper in the bloodstream. People with Wilson disease often have low ceruloplasmin levels, but not always.
- copper. People with Wilson disease may have lower than normal blood copper levels. Acute liver failure due to Wilson disease may cause high blood copper levels.
- liver enzymes alanine transaminase (ALT) and aspartate transaminase (AST). People with Wilson disease may have abnormal ALT and AST levels.
- red blood cells to look for signs of anemia.
Doctors may order a blood test to check for the gene mutations that cause Wilson disease if other medical tests don’t confirm or rule out a diagnosis of the disease.
24-hour urine collection test
For 24 hours, you will collect your urine at home in a special container that is copper-free, provided by a health care professional. A health care professional will send the urine to a lab, which will check the amount of copper in your urine. Copper levels in the urine are often higher than normal in people who have Wilson disease.
Liver biopsy
If the results of blood and urine tests don’t confirm or rule out a diagnosis of Wilson disease, your doctor may order a liver biopsy. During a liver biopsy, a doctor will take small pieces of tissue from your liver. A pathologist will examine the tissue under a microscope to look for features of specific liver diseases, such as Wilson disease, and check for liver damage and cirrhosis. A piece of liver tissue will be sent to a lab, which will check the amount of copper in the tissue.
Imaging tests
In people who have nervous system symptoms, doctors may use imaging tests to check for signs of Wilson disease or other conditions in the brain. Doctors may use
- magnetic resonance imaging (MRI), which uses radio waves and magnets to produce detailed images of organs and soft tissues without using x-rays
- computed tomography (CT) scan, which uses a combination of x-rays and computer technology to create images
Treatment
How do doctors treat Wilson disease?
Doctors treat Wilson disease with
- medicines that remove copper from the body, called chelating agents
- zinc, which prevents the intestines from absorbing copper
In many cases, treatment can improve or prevent symptoms and organ damage. Doctors may also recommend changing your diet to avoid foods that are high in copper.
People who have Wilson disease need lifelong treatment. Stopping treatment may cause acute liver failure. Doctors regularly perform blood and urine tests to check how the treatment is working.
Chelating agents
Penicillamine (Cupramine, Depen) and trientine (Syprine) are two chelating agents used to treat Wilson disease. These medicines remove copper from the body.
Penicillamine is more likely to cause side effects than trientine. Side effects of penicillamine may include fever, rash, kidney problems, or bone marrow problems. Penicillamine may also reduce the activity of vitamin B6, and doctors may recommend taking a vitamin B6 supplement along with penicillamine. In some cases, when people with nervous system symptoms begin taking chelating agents, their symptoms get worse.
When treatment begins, doctors gradually increase the dose of chelating agents. People take higher doses of chelating agents until the extra copper in the body has been removed. When Wilson disease symptoms have improved and tests show that copper is at safe levels, doctors may prescribe lower doses of chelating agents as maintenance treatment. Lifelong maintenance treatment prevents copper from building up again.
Chelating agents may interfere with wound healing, and doctors may prescribe a lower dose of chelating agents for people who are planning to have surgery.
Zinc
Zinc prevents the intestines from absorbing copper. Doctors may prescribe zinc as a maintenance treatment, after chelating agents have removed extra copper from the body. Doctors may also prescribe zinc for people who have Wilson disease but do not yet have symptoms. The most common side effect of zinc is stomach upset.
How do doctors treat Wilson disease in women who are pregnant?
Pregnant women should continue treatment for Wilson disease throughout pregnancy. Doctors may prescribe a lower dose of chelating agents for women who are pregnant. Since the fetus needs a small amount of copper, lowering the dose may keep copper at safe levels without removing too much copper.
In most cases, doctors recommend that women continue to take the full dose of zinc during pregnancy. Experts recommend that women with Wilson disease do not breastfeed if they are taking chelating agents. Penicillamine is present in breast milk and can be harmful to a baby. Experts have little information about the safety of trientine and zinc in breast milk.
How do doctors treat the complications of Wilson disease?
If Wilson disease leads to cirrhosis, doctors can treat health problems and complications related to cirrhosis with medicines, surgery, and other medical procedures.
If Wilson disease causes acute liver failure or liver failure due to cirrhosis, you may need a liver transplant. A liver transplant cures Wilson disease in most cases.
Can I prevent Wilson disease?
You can’t prevent Wilson disease. If you have a first-degree relative—a parent, sibling, or child—with Wilson disease, talk with your doctor about testing you and other family members for the disease. A doctor may be able to diagnose and begin treating Wilson disease before symptoms appear. Early diagnosis and treatment can reduce or prevent organ damage.

Eating, Diet, & Nutrition
What should I avoid eating if I have Wilson disease?
When you start treatment for Wilson disease, your doctor may recommend avoiding foods that are high in copper, such as
- chocolate
- liver
- mushrooms
- nuts
- shellfish
After treatments have lowered your copper levels and you begin maintenance treatment, talk with your doctor about whether you can safely eat moderate amounts of these foods.
If your tap water comes from a well or runs through copper pipes, have the copper levels in your water checked. Water sitting in copper pipes may pick up copper. Run the water to flush the pipes before you drink the water or use it for cooking. You may need to use a water filter to remove copper from your tap water.
For safety reasons, talk with your doctor before using dietary supplements, such as vitamins, or any complementary or alternative medicines or medical practices. Some dietary supplements may contain copper.

Clinical Trials
The National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and other components of the National Institutes of Health (NIH) conduct and support research into many diseases and conditions, including liver diseases.
What are clinical trials for Wilson disease?
Clinical trials—and other types of clinical studies—are part of medical research and involve people like you. When you volunteer to take part in a clinical study, you help doctors and researchers learn more about disease and improve health care for people in the future.
Researchers are conducting clinical studies to better understand liver diseases, such as Wilson disease.
Find out if clinical studies are right for you.
Watch a video of NIDDK Director Dr. Griffin P. Rodgers explaining the importance of participating in clinical trials.
What clinical studies for Wilson disease are looking for participants?
You can view a filtered list of clinical studies on Wilson disease that are federally funded, open, and recruiting at ClinicalTrials.gov. You can expand or narrow the list to include clinical studies from industry, universities, and individuals; however, the NIH does not review these studies and cannot ensure they are safe. Always talk with your health care provider before you participate in a clinical study.
How is NIDDK- and NIH-funded research advancing the understanding of Wilson disease?
The NIDDK and other components of the NIH support basic research to increase our understanding of Wilson disease and lay the foundation for future advances in diagnosis and treatment. Research topics include
- understanding how copper is absorbed from the intestines, handled by tissues including brain and liver tissues, and then removed from the body
- understanding how the gene mutations in Wilson disease lead to copper not being properly removed and building up in the brain, liver, red blood cells, and kidneys
- developing better tests that doctors could use to check for Wilson disease in all infants at birth
- developing better treatments for Wilson disease and better ways to check copper levels in the blood and liver
- developing gene therapy for Wilson disease that might keep copper in the body at safe levels without the need for a special diet or lifelong treatment with chelating agents
This content is provided as a service of the National Institute of Diabetes and Digestive and Kidney Diseases
(NIDDK), part of the National Institutes of Health. NIDDK translates and disseminates research findings to increase knowledge and understanding about health and disease among patients, health professionals, and the public. Content produced by NIDDK is carefully reviewed by NIDDK scientists and other experts.
The NIDDK would like to thank:
Valentina Medici, M.D., University of California, Davis